М’язова слабкість, яка краде перші кроки дитини, задишка, що приходить непрошено вночі, серце, що б’ється нерівно під вагою накопиченого глікогену, — ось реальність хвороби Помпе. Це рідкісний генетичний розлад, глікогеноз II типу, коли дефіцит ферменту кислої альфа-глюкозидази (GAA) перетворює клітини м’язів на склади непотрібного цукру. Без лікування немовлята не доживають до двох років, а дорослі борються з прогресуючою втомою, але ферментозамісна терапія (ФЗТ) вже дарує роки повноцінного життя. В Україні МОЗ закуповує препарати, як алглюкозидаза альфа, роблячи надію ближчою.
Уявіть м’язи, які мали б бути сильними, як стрункі мотузки, а стали млявими від “глікогенового сміття” в лізосомах. Хвороба вражає скелетні м’язи, діафрагму, серце — скрізь, де потрібен рух. Частота — один випадок на 40 тисяч новонароджених у Європі, але в Україні діагностують десятки, часто запізно. Раннє скринінгування новонароджених і генетичні тести змінюють гру, дозволяючи почати ФЗТ до незворотних змін.
Ключ до порятунку — вчасна дія. Симптоми починаються по-різному: у немовлят гіпотонія та збільшене серце, у дорослих — втома при сходах і апное уві сні. Сучасні препарати на кшталт Nexviazyme чи Pombiliti перевершують перші покоління, проникаючи глибше в м’язи. Давайте розберемося детально, щоб ви могли розпізнати сигнали і боротися розумно.
Що ховається за назвою: суть хвороби Помпе
Глікоген — паливо для м’язів, але в лізосомах його розщеплює GAA-фермент. Мутація в гені GAA на 17-й хромосомі блокує цей процес, і глікоген накопичується, руйнуючи клітини зсередини. Це не просто слабкість: лізосоми розриваються, вивільняючи токсини, м’язи атрофуються, серце перевантажується. Хвороба Помпе — лізосомна хвороба накопичення, одна з небагатьох, де ФЗТ працює драматично.
Історія починається з 1932 року, коли Йоганнес Помпе описав немовля з гіпертрофією серця. Сьогодні знаємо понад 600 мутацій GAA — від повної відсутності ферменту в інфантильній формі до часткової в дорослій. Аутосомно-рецесивне успадкування означає: обидва батьки — носії, ризик для дитини 25%. Ви не повірите, але в деяких популяціях, як афроамериканців чи тайванців, частота вища через “засновницькі” мутації.
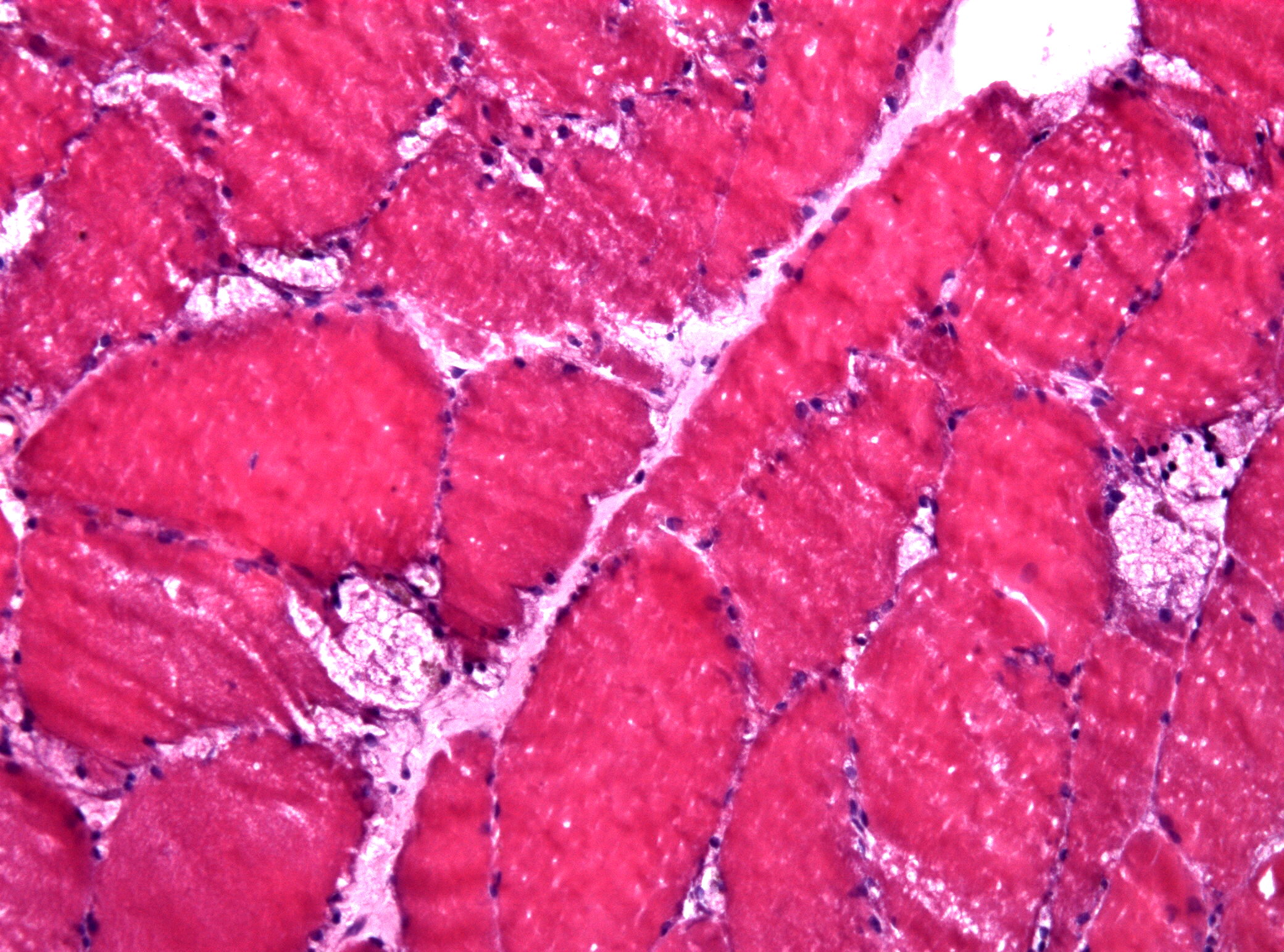
Патогенез хитрий: глікоген не просто лежить — він порушує автоліз, викликає запалення, фіброз. У м’язах з’являються вакуолі, серце гіпертрофується. Без ФЗТ прогрес нестримний, але терапія розчиняє накопичення, відновлюючи функцію. Ранній старт ФЗТ у немовлят подовжує життя з 1 року до нормального.
Форми хвороби Помпе: від блискавичної до повільної
Хвороба не монолітна — спектр від класичної інфантильної (IOPD) до晚-onset (LOPD). Класична IOPD дебютує до 3 місяців: дитина “лялькова”, голова не тримається, живіт випирає від гепатомегалії, серце величезне. Без лікування — смерть від серцевої чи дихальної недостатності до 2 років. Non-classic infantile — між 3 і 12 місяцями, менше кардіо, більше м’язова слабкість.
LOPD — найпоширеніша, 90% випадків, починається після року, часто в 20-50. Тут серце рідко страждає, зате діафрагма слабне, з’являється “качина хода”, сколіоз. Прогрес повільний, хворий може ходити роками, але вентиляція легень стає невід’ємною. Переходи плавні: симптоми залежать від рівня GAA — менше 1% у IOPD, 1-30% у LOPD.
| Форма | Вік дебюту | Основні ураження | Прогноз без лікування |
|---|---|---|---|
| Класична інфантильна (IOPD) | <3 міс | Серце, м’язи, дихання | Смерть <2 років |
| Non-classic infantile | 3-12 міс | М’язи, дихання, менше серце | Смерть в ранньому дитинстві |
| Późній початок (LOPD) | >1 рік | Скелетні м’язи, діафрагма | Респіраторна недостатність у 20-50 років |
Таблиця базується на даних rarediseases.org. Порівняння показує, чому скринінг новонароджених — ключ: у США та Тайвані виявляють IOPD одразу.
Симптоми хвороби Помпе: тривожні сигнали
У немовлят перше — гіпотонія: дитина не стискає кулачки, не перевертається, смокче слабо. Серце чути здалеку — гіпертрофічна кардіоміопатія, ЕКГ показує короткий PR-інтервал. Макроглосія робить рот напіввідкритим, дихання шумне від слабкої діафрагми. Батьки помічають: “Він не набирає вагу, кашляє ночами”. У дітей старше — затримка ходьби, часті пневмонії, біль у м’язах після навантаження.
Дорослі ігнорують роками: тяжкість у ногах при сходах, вставання з підлоги руками (симптом Говерса), сонливість вдень від апное. Жінки частіше мають слабкість плечового поясу, чоловіки — тазового. Додатково: рефлюкс, запори, птоз повік, сколіоз як наслідок слабких спинних м’язів. У 70% LOPD пацієнтів дихальна недостатність — головна причина інвалідності.
- Ранні ознаки у немовлят: провисання голови, слабкий плач, збільшений язик, задишка в спокої.
- У дітей: качина хода, труднощі з бігом, часті ГРВІ з ускладненнями.
- У дорослих: втома після роботи, головний біль вранці від гіпоксії, потреба в опорі при вставанні.
Ці пункти — чек-лист для лікарів. Часто плутають з ДЦП чи міастенією, але високий КФК і вакуолі в біопсії видають Помпе.
Діагностика хвороби Помпе: від підозри до підтвердження
Підозра виникає за клінікою: проксимальна слабкість + респіраторні проблеми + родинний анамнез. Перший крок — суха пляма крові на GAA-активність (чутливість 95%). Низький рівень — генетичний тест на мутації GAA. У фібробластах точніше для LOPD. Біопсія м’язів: глікогенові вакуолі під мікроскопом — класика.
Інструментал: ЕхоКГ для кардіоміопатії, СКГ сонна для апное, МРТ м’язів для атрофії. В Україні — тести в Охматдиті, Львові (з 2024 для Помпе, Фабрі). Новонароджений скринінг вводять поступово. Час критичний: діагноз за тижні рятує серце в IOPD.
- Збір анамнезу та огляд невролога.
- Біохімія: КФК↑, АЛТ/АСТ↑, GAA↓.
- Генетика: секвенування GAA.
- Підтвердження: біопсія чи пренатал.
Після списку — мультидисциплінарний підхід: генетик, кардіолог, пульмонолог. Успіх — у команді.
Сучасне лікування хвороби Помпе: від ФЗТ до генної терапії
ФЗТ — золото стандарту з 2006. Alglucosidase alfa (Myozyme) вводять кожні 2 тижні в/в, розчищаючи глікоген. У IOPD виживає 90%+ за 5 років. Новіші: Nexviazyme (2021, avalglucosidase alfa) — вдвічі ефективніший у м’язах завдяки маннозо-6-фосфату. Pombiliti + Opfolda (2023) для LOPD дорослих — фермент + шаперон, покращує дихання на 20%.
В Україні МОЗ закуповує алглюкозидазу альфа (2025), Охматдит лікує дітей безкоштовно. Підтримка: НІВЛ (неінвазивна вентиляція) від апное, фізіо для м’язів, дієта багата протеїном, ортези. Майбутнє — генна терапія: AB-1009 (AskBio, фаза 1/2 2026) доставляє GAA AAV-вектором до печінки, потенційно одноразово. Тріали обіцяють революцію.
Дози ФЗТ ростуть з вагою: 20 мг/кг для Myozyme, 30 для Nexviazyme. Побочки: алергія (5%), антитіла знижують ефект — моніторинг ключовий. Комбінація з модafinілом для сонливості — хитрість практиків.
Поради для сімей з хворобою Помпе
Не ігноруйте втомленість — ведіть щоденник симптомів для лікаря. Почніть фізіо рано: плавання зміцнює м’язи без навантаження. Вакцинуйте від пневмококів — інфекції фатальні. Шукайте групи підтримки: в Україні Асоціація Помпе, форуми rare-diseases.com.ua. Плануйте генконсультацію — для братів/сестер ризик 1-2%. Харчуйтеся калорійно: смузі з протеїном рятують вагу. І головне — святкуйте малі перемоги, бо з ФЗТ життя яскраве!
Прогноз при хворобі Помпе та якості життя
Без лікування IOPD — трагедія, LOPD — інвалідність у 40-50. З ФЗТ немовлята ходять, дихають самостійно; дорослі повертаються до роботи. У кейсі з Охматдиту дівчинка Софійка на ФЗТ бігає в садок. Статистика: виживання IOPD з 0% до 75% за 10 років. Виклики лишаються: імуногенність, доступність в Україні (черги на ліки).
Якість життя — в деталях: НІВЛ ночі звільняє від маски вдень, ортопедія виправляє поставу. Психологічна допомога родинам — must, бо стрес посилює симптоми. Тренди 2026: генна терапія в клініках, скринінг по всій Україні. Хвороба Помпе більше не вирок — це виклик, який приймають з оптимізмом.
Кожен день на ФЗТ — крок до нормальності. Сучасні препарати, як Nexviazyme, проникають туди, де попередники не могли, даруючи силу. Батьки, лікарі, пацієнти — разом ми перемагаємо накопичення, повертаючи м’язам свободу. Розмова про Помпе триває: нові тріали, історії успіху надихають на бій.